
Amyotrophic Lateral Sclerosis (ALS)
Many, if not most, neurodegenerative diseases are characterized by the conversion of native folded protein to a misfolded form via protein templating. Amyotrophic Lateral Sclerosis (ALS) has sometimes been referred to as “Lou Gehrig’s disease” and is a progressive and fatal neurodegenerative disease that is characterized by progressive degeneration of motor neurons in the brain and spinal cord. Approximately 95% of ALS is sporadic, with proteins such as Transactivation Response DNA binding protein of 43 kiloDalton (TDP-43) implicated in sporadic ALS (sALS) whereas, Copper and Zinc Superoxide Dismutase (Cu/Zn SOD) has been shown to be involved in SOD familial ALS (fALS). Below, we provide information from:
https://www.ninds.nih.gov/health-information/disorders/amyotrophic-lateral-sclerosis-als
What is amyotrophic lateral sclerosis (ALS)?
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's Disease, is a rare neurological disease that affects motor neurons—those nerve cells in the brain and spinal cord that control voluntary muscle movement. Voluntary muscles are those we choose to move to produce movements like chewing, walking, and talking. The disease is progressive, meaning the symptoms get worse over time. ALS has no cure and there is no effective treatment to reverse its progression.
ALS is a type of motor neuron disease. As motor neurons degenerate and die, they stop sending messages to the muscles, which causes the muscles to weaken, start to twitch (fasciculations), and waste away (atrophy). Eventually, the brain loses its ability to initiate and control voluntary movements.
Early symptoms include:
Muscle twitches in the arm, leg, shoulder, or tongue
Muscle cramps
Tight and stiff muscles (spasticity)
Muscle weakness affecting an arm, a leg, the neck, or diaphragm
Slurred and nasal speech
Difficulty chewing or swallowing
As the disease progresses, muscle weakness and atrophy spread to other parts of your body. You may develop problems with:
People with ALS eventually will not be able to stand or walk, get in or out of bed on their own, or use their hands and arms
Chewing food and swallowing (dysphagia)
Speaking or forming words (dysarthria)
Breathing (dyspnea); individuals with ALS eventually lose the ability to breathe on their own and must depend on a ventilator
Maintaining weight and malnourishment
Muscle cramps and neuropathy (nerve damage or disease)
Anxiety and depression, because people with ALS usually remain able to reason, remember, understand, and are aware of their progressive loss of function
Although not as common, people with ALS may also:
Experience problems with language or decision-making
Develop a form of dementia over time
ALS doesn't affect your ability to taste, touch, or smell, or hear. Most people with ALS die from respiratory failure, usually within three to five years from when the symptoms first appear. However, about 10 percent of people with ALS survive for a decade or more.
Who is more likely to get amyotrophic lateral sclerosis (ALS)?
Risk factors for ALS include:
Age—Although the disease can strike at any age, symptoms most commonly develop between the ages of 55 and 75.
Biological sex—Males are slightly more likely to develop ALS. However, as people age the difference between the sexes disappears.
Race and ethnicity—Caucasians and non-Hispanics are most likely to develop the disease, but ALS affects people of all races and ethnic backgrounds.
Some studies suggest that military veterans are about one and half to two times more likely to develop ALS, although the reason for this is unclear. Possible risk factors for veterans include exposure to lead, pesticides, and other environmental toxins.
Sporadic and Familial ALS
Nearly all cases of ALS are considered sporadic. This means the disease seems to occur at random with no clearly associated risk factors and no family history of the disease. Although family members of people with sporadic ALS are at an increased risk for the disease, the overall risk is very low and most will not develop ALS.
About five to 10 percent of all ALS cases are familial (also called inherited or genetic). Mutations in more than a dozen genes have been found to cause familial ALS.
About 25 to 40 percent of all familial cases (and a small percentage of sporadic cases) are caused by a defect in the C9ORF72 gene, which makes a protein that is found in motor neurons and nerve cells in the brain. Some people with this gene also develop a type of frontotemporal degeneration (FTD, a form of dementia) caused by atrophy of the brain's temporal and frontal lobes.
Another 12 to 20 percent of familial cases result from mutations in the SOD1 gene that is involved in production of the enzyme copper-zinc superoxide dismutase 1.
In 2021, a team of scientists led by the NIH and the Uniformed Services University of the Health Sciences announced it had discovered a unique form of genetic ALS that affects children as early as age 4 years. This childhood form of ALS is linked to the gene SPTLC1 that is part of the body's fat production system and may be caused by changes in the way the body metabolizes fatty materials called lipids.
How is amyotrophic lateral sclerosis (ALS) diagnosed and treated?
Diagnosing ALS
There is no single test that can definitely diagnose ALS. Your healthcare provider will conduct a physical exam and review your full medical history. A neurologic examination will test your reflexes, muscle strength, and other responses and will be held at regular intervals to assess whether symptoms such as muscle weakness, muscle wasting, and spasticity are progressively getting worse.
Muscle and imaging tests to rule out other diseases and confirm the diagnosis include:
Electromyography (EMG) is a recording technique that detects electrical activity of muscle fibers and can help diagnose ALS.
A nerve conduction study (NCS) measures the electrical activity of your nerves and muscles by assessing the nerve's ability to send a signal along the nerve or to the muscle.
Magnetic resonance imaging (MRI) is a noninvasive procedure that uses a magnetic field and radio waves to produce detailed images of the brain and spinal cord.
Blood and urine tests may be performed based on your symptoms, test results, and findings from the examination by a doctor. A physician may order these tests to eliminate the possibility of other diseases.
A muscle biopsy may be performed if your doctor believes you may have a muscle disease other than ALS. Under local anesthesia, a small sample of muscle is removed and sent to the lab for analysis.
Treating ALS
There is no treatment to reverse damage to motor neurons or cure ALS. However, treatments can make living with the disease easier. See the website below for more information:
https://www.ninds.nih.gov/health-information/disorders/amyotrophic-lateral-sclerosis-als
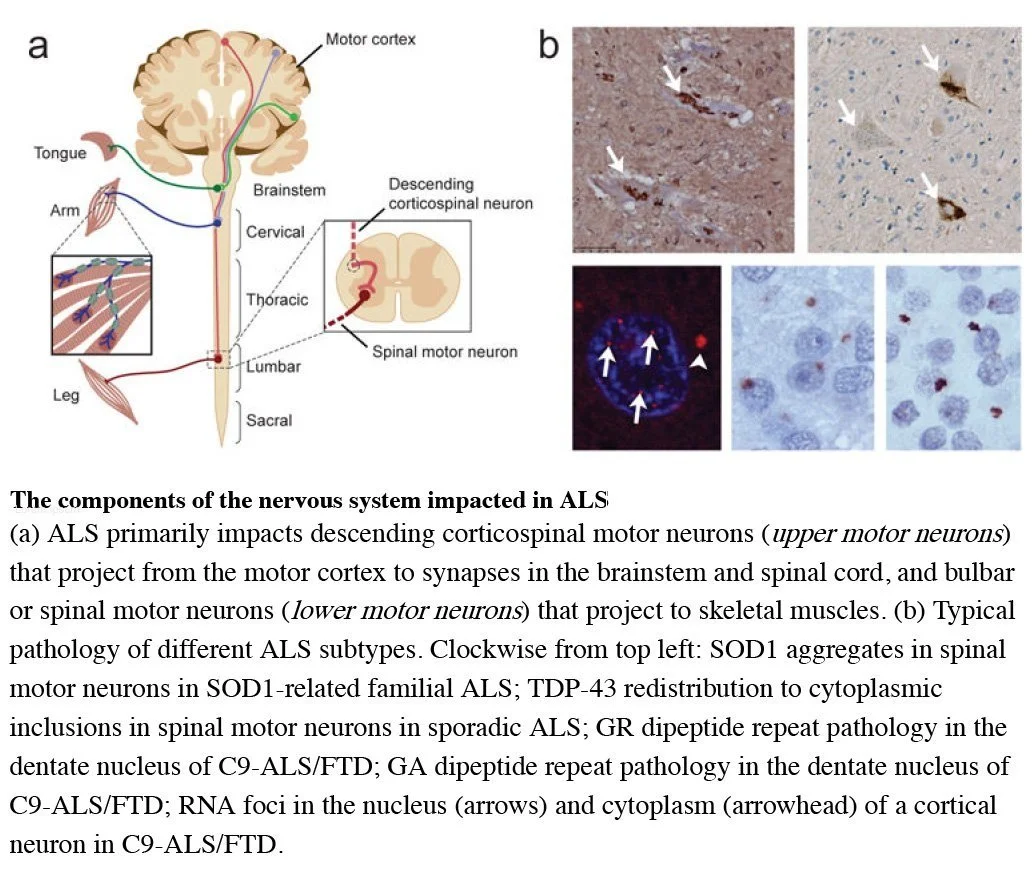
From JP Taylor, RH Brown, Jr and DW Cleveland (2016) Decoding ALS: From genes to mechanism. Nature 539, 197–206. doi:10.1038/nature20413.